EU-MDR
What is EU-MDR compliance?
The European Medical Device Regulation (MDR) is a set of regulations that governs the production and distribution of medical devices in Europe, and compliance with the regulation is mandatory for medical device companies that want to sell their products in the European marketplace. Manufacturers must develop a suite of MDR-compliant regulatory systems, processes, and documents to continually monitor the safety and performance of their products.
Europe
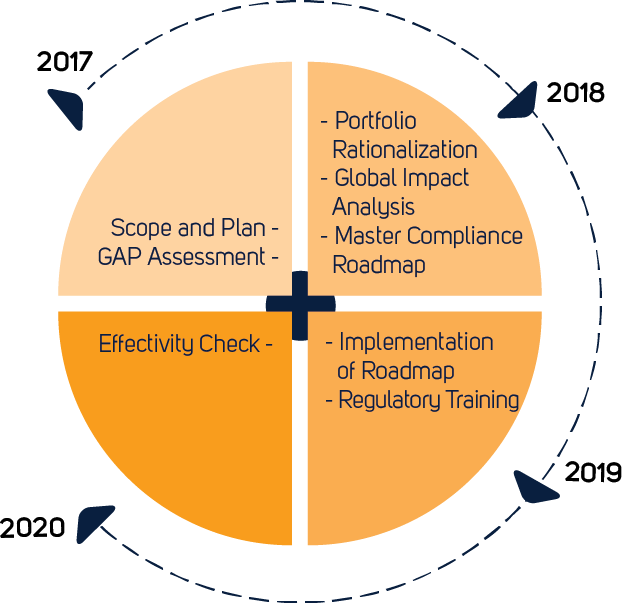
From MDD to MDR in 2021
The Medical Devices Regulation (EU 2017/745) has replaced the Medical Devices Directive (93/42/EEC) as the legislation detailing the requirements that manufacturers must meet to place medical devices on the market in the European Union. All manufacturers of Class I to III medical products must familiarize themselves with the new requirements as soon as possible. The regulation has far-reaching implications and affects all classes of medical products.
Compared with the MDD, the new European MDR is less focused on the pre-approval stage of medical device manufacturing, and instead, promotes a life-cycle approach to medical device regulation. Successful MDR compliance is demonstrated by the application of a CE-mark to the device. Class I manufacturers, except for devices that are sterile or have a measuring function, may self-apply a CE-mark after producing a declaration of conformity. For all other classes of medical device, a CE-mark may only be affixed once a Notified Body has issued a certificate of conformity following a regulatory review according to rules in Chapter IV of the MDR.
Key changes you want to know about
Product scope expansion - The definition of medical devices and active implantable medical devices covered under the MDR will be significantly expanded to include devices that have not a medical intended purpose.
Identification of "qualified person" - Device manufacturers will be required to identify at least one person within their organization who is ultimately responsible for all aspects of compliance with the requirements of the new regulation.
Implementation of unique device identification - The MDR mandates the use of unique device identification (UDI) mechanisms. This requirement is expected to increase the ability of manufacturers and Authorities to trace specific devices through the supply chain.
Reclassification of devices according to risk, contact duration, and invasiveness - The MDR will require device manufacturers to review the updated classification rules and update their technical documentation accordingly by considering the fact that class III and implantable devices will have higher clinical requirements and a regular scrutiny process.
Clinical evidence for class III and implantable medical devices - Manufacturers will need to conduct clinical investigations in case they do not have sufficient clinical evidence to support the claims done on both safety and performance of a dedicated device.
Clinical evaluation of Class IIa and Class IIb medical devices - Manufacturer will need to re-prepare their clinical evaluation by considering the new wording of the regulation on when an equivalence approach and under which circumstances it is possible to justify not conducting a clinical investigation.
Post-market oversight - The MDR request increased post-market surveillance authority by the Notified Body.
No "grandfathering" provisions - Under the MDR, all currently approved devices must be recertified in accordance with the new requirements.
Europe
Get trained on hot EU-MDR topics by our Experts
Qserve has designed a 11-session virtual training program to assist manufacturers at every stage & step of implementation to align your company with the new requirements of the Medical Device Regulation (EU-MDR).
The program includes exclusive content developed by Qserve's experts and is suitable for professionals at any level of knowledge. Don't miss this opportunity. This MDR Suite will start February 2022.
All MDR Suite courses