
Our mission is to help improve patient safety and health by supporting the advancement of biomedical technology. We assist medical device and in-vitro diagnostics manufacturers worldwide with market access, medical device approvals, clinical trials and achieving and maintaining regulatory compliance.
We strive to do this as your partner with a practical approach balancing business needs and regulatory compliance.
We are passionately working for our customers on regulatory, quality, and clinical challenges and see it as our responsibility to create a diverse global team of professionals who share the same passion for medical device technology, a high standard of quality, competence, and customer service, and enjoy the fun of working on this together every day. Our people make our firm.

Our Therapeutic Specialties
We are widely recognized for our"practical" mindset—many of our consultants come from the MedTech industry, often having worked in regulatory, quality, clinical, or R&D roles or having been employed by notified bodies. This makes their advice practical, implementation-ready, and grounded in operational reality, not just theoretical knowledge.




The board of Qserve Group
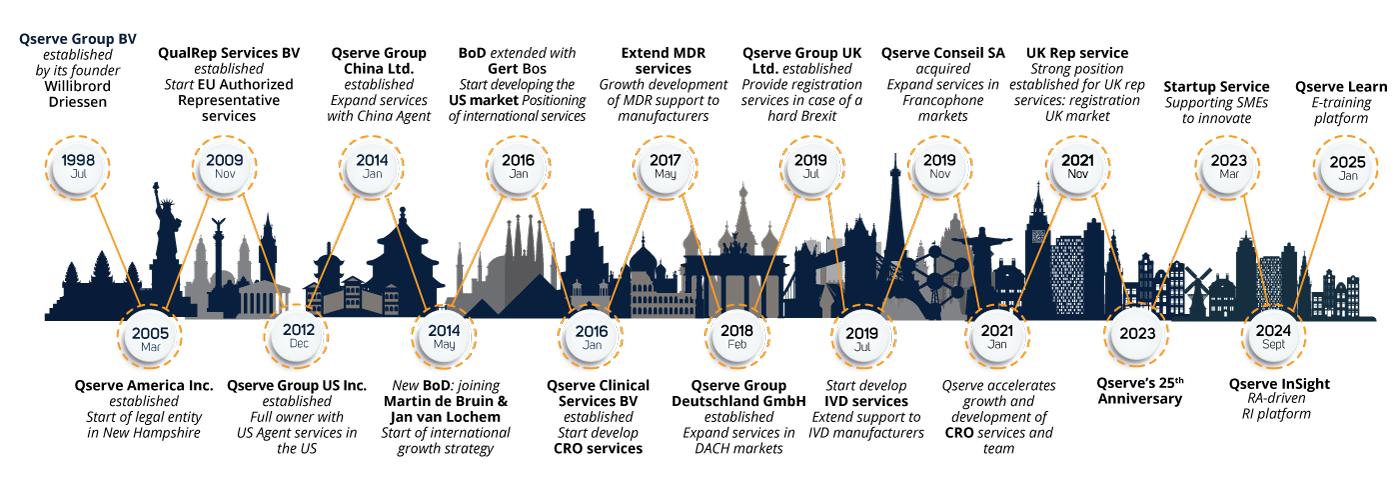
Qserve Group was founded in 1998 by Willibrord Driessen with a small team of regulatory experts. Since 2014, three new shareholders joined the Board of Directors, sharing Willibrord's global vision. Today, Qserve is a well-known global player in the medical device industry, with offices in Europe, the United States, and China, serving manufacturers worldwide.
The Board of Qserve Group includes Jan van Lochem, Martin de Bruin, and Gert Bos. They are committed to expanding Qserve Group and Qserve CRO into a top global medical device consulting group. We specialize in regulatory and clinical affairs, Regulatory Intelligence, Quality Assurance, Clinical Research and Training to help medical devices and diagnostics gain registration, approval, and market access worldwide.

Over the past 27 years, Qserve has developed into a global company where the team combines their regulatory knowledge and experience in the medical device and in vitro diagnostic industry, sharing more than 500 years' worth of combined expertise in the medical field.
The historical development shows Qserve makes you your global partner for Regulatory Compliance all over the world.
We have 7 offices worldwide







Meet our team of professionals

Jan van Lochem, MSc, MBA
CEO
Partner

Martin de Bruin, MSc
CCO
Partner

Gert Bos, PhD
CSO
Partner
.png)
Amé Huige
Marketing Specialist
Manager Training Marketing & Development

Dulce Aguilar, PhD
Clinical Writer

Jorn van Binsbergen, MSc
Clinical Writer

Claire Borde, PhD
Medical Writer

Renaud Brendel
Country Manager
Senior Consultant

Maria Cámara Torres, PhD
Medical Writer

Minghua Chen
Training Expert
MDR Expert

Mindy McCann
Country Manager
VP US Operations

Patrícia da Silva Perez, PhD
Team Manager Medical Writing
Senior Consultant

Coenraad Davidsdochter, MSc
Team Manager Electrical Safety and Software/AI

Nienke Flipsen-Maassen, MSc
Data Manager

Nicolas Garrett
IVD Regulatory Affairs Expert

Adriana Gavrilciuc, MSc
Regulatory Affairs Expert

Olena Hoi, MSc
Quality and Regulatory Expert
%20Huang.png)
Stephanie (Xing) Huang
Country Manager
Principal Consultant
NMPA Expert
Clinical Study Expert

Jasmin Hunter
Clinical and Regulatory Affairs Expert
Floor Janssen
Medical Writer

Tim Joiner, MSc
Quality and Auditing Expert

Kevin Kenney
Electromechanical Design and Safety Compliance Expert

Jelena Kresoja, MSc
Clinical Writer
Jiuru Li
Medical Writer
Bingshuo Li, PhD
Active Medical Devices Expert
Dr. Cornelia Luban, PhD
Regulatory Affairs Expert

Bianca Lutters, PhD
Head of Clinical Operations

Wouter Mattheussens, MSc
Clinical Project Manager

Keith Morel, PhD
VP US Regulatory Compliance
Principal Consultant

Henk-Willem Mutsaers, MSc
Principal Consultant
Quality Assurance and Auditing Expert

François Naye, PhD
Senior Consultant
Training Expert
Software Expert
Quality Expert

Inette Nieveen, MSc
Head of Medical Devices

Robert Paassen, MSc
Regulatory Affairs Expert
Quality Expert
Swaroop Rani Kesevan Nair
Consultant
Cécile Rosset
Senior Consultant

Dennis Sarwin, MSc

René Schings
Quality and Auditing Expert
Principal Consultant
Training Expert

Kristiane Schmidt, PhD
Senior Consultant

Agnieszka Schreiber, BEng
Senior Consultant
Jennifer Simon, MSc
Regulatory Affairs Expert